以前紹介した「医薬品の特許紛争の早期解決システムの実施規則(药品专利纠纷早期解决机制实施办法)(試行)」の最終版が公表され、7月4日に施行されました。続いて、それに関連する裁判所、特許庁の細則も7月5日に公表されて施行され本格的に動き出すことになりました。
パテントリンケージ制度の全体像
従来、ジェネリック企業があるジェネリック薬の上市の承認申請をした場合、中国の薬事当局はその審査段階においてジェネリック薬が医薬品として必要な基準を満たしているか否かの審査をするだけではなく、特許に関連する問題も判断していました。つまり、当該ジェネリック薬の製造・販売が対応の新薬(先発品)をカバーする新薬特許を侵害するか否かについてもある程度のレベルで判断がされていて、問題がないと判断されてはじめてそのジェネリック薬に対して上市の承認が付与されていました。そこには外部の様々な利害関係者からの圧力がかかっていたであろうことは想像に難くありません。そして今回のパテントリンケージ制度では、特許問題の判断にあたっては薬事当局がジェネリック薬の承認審査段階で単独で行うのでなく、裁判所、特許局とも緊密な連携を取って早期に解決しようというものです。
したがって特許法76条に定められているパテントリンケージ制度については、上記の3つの機関がそれぞれ下記の通り細則・弁法等を公表した上で施行されています。
これらの弁法・規定を引用しながら、下記の全体像に従って説明を進めて行きます。
特許情報プラットフォームの開設
中国の薬事当局である医薬品監督管理局(NMPA)の審査センター(CDE)がパテントリンケージ制度の肝となる、新薬とそれにリンクした特許を収録した特許情報プラットフォーム(https://zldj.cde.org.cn/home)を開設しその管理責任を負います(パテントリンケージシステムの実施弁法 §2,3)。特許情報プラットフォームはアメリカのオレンジブックの中国版と言えます。
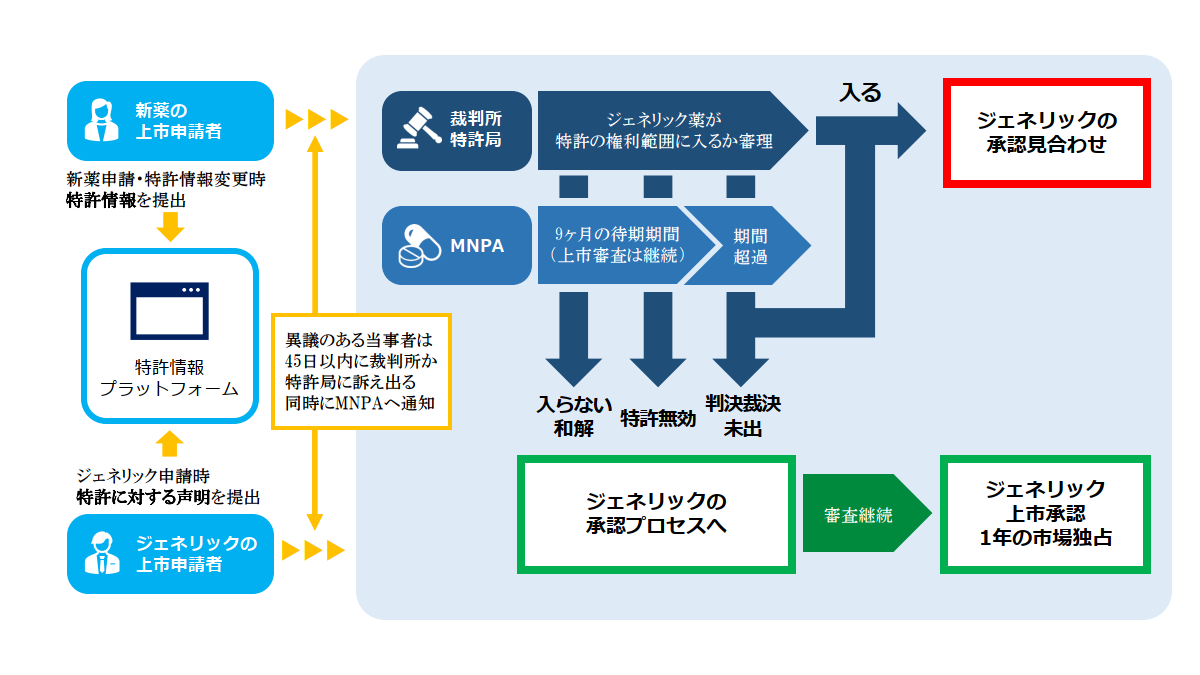
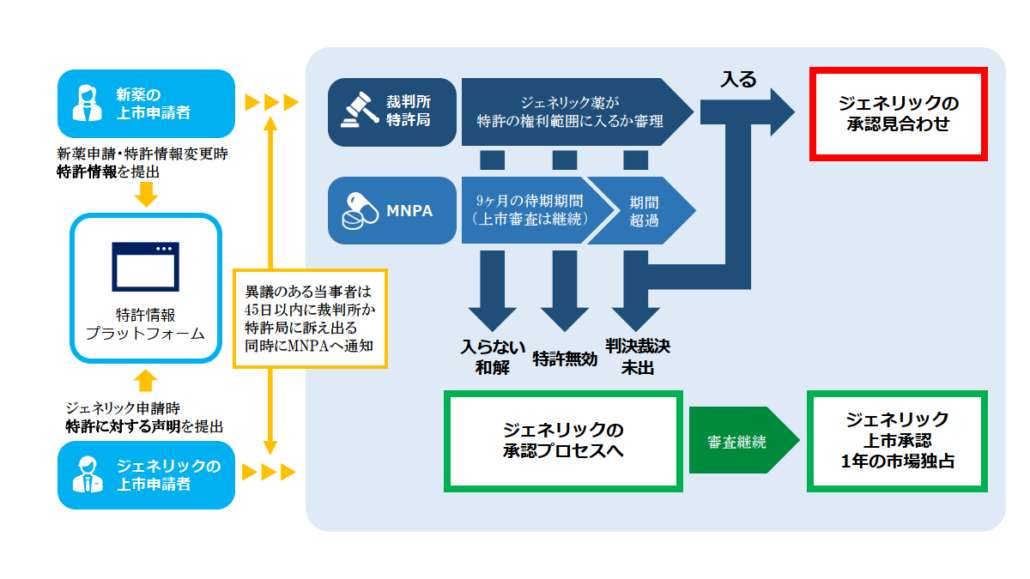
パテントリンケージ制度でのプロセスは以下の通りです。
①新薬の特許情報の提供
新薬の特許情報の登録
新薬を開発した企業はNMPAに上市承認の申請をし審査を受けた結果として承認を取得した場合には、当該新薬をカバーする特許(新薬特許)を特許情報プラットフォームに入力します。この入力は新薬の上市承認を取得した者(新薬承認取得者)が承認取得後30日以内に行う必要があります。登録すべき事項には下記が含まれます。
新薬の名称、剤型、規格、新薬承認取得者、関連特許番号、特許名称、特許権者、ライセンシー、特許成立日、特許満了日、成立状態、特許の類型(物質、用途、製剤等)、承認対象の新薬と当該新薬をカバーする特許クレーム(新薬特許クレーム)との関係、連絡先・その住所、連絡方法等。
パテントリンケージシステムの実施弁法 §4
前述の通り、特許情報プラットフォームの管理責任はCDEが負いますが、登録された情報の正確性や完全性等は申請人の新薬承認取得者が責任を負います(パテントリンケージシステムの実施弁法 §4)。内容に変更があった場合には30日以内に特許情報プラットフォームに登録しなければなりません(パテントリンケージシステムの実施弁法 §4)。
このように登録された情報は、公衆に公開されます。
登録・公開された情報について第三者は下記の点で異議を申し立てることができ、新薬承認取得者は必要に応じて特許情報プラットフォームの情報を修正し、その修正理由についても公表する必要があります。
特許情報プラットフォーム掲載された情報と特許簿、特許公報、販売承認書の情報の不一致
新薬特許に含まれる用途特許と新薬承認対象の能書の適用症が不一致
新薬特許クレームが上市承認対象の新薬の対応技術をカバーしていない
パテントリンケージシステムの実施弁法 §4
対象となる新薬特許の類型(パテントリンケージシステムの実施弁法 §5,12)
中国のパテントリンケージ制度の対象となる新薬の特許の類型は下記の通りです。
- 低分子医薬:物質特許、製剤特許、医薬用途特許
- バイオ医薬:配列特許、医薬用途特許
- 漢方薬:組成物・製剤特許、抽出物特許、用途特許
したがって本パテントリンケージ制度の下で審理の対象になるのは、新薬について上記の類型の特許であり、特許情報プラットフォームに登録されている特許に基づいてなされる提訴・請求のみです(パテントリンケージシステムの実施弁法 §2、最高裁規定 §2、特許局弁法 §4(4))。
上記以外の特許の類型(例えば化合物の製造方法)はパテントリンケージ制度の対象特許とはなりません。ですから対象外の類型の特許に基づいてパテントリンケージ制度の下で提訴しても、裁判所はこれを却下します(特許法 §76)。認められていない類型の特許に基づくジェネリック薬に対する差し止め訴訟は、通常の侵害訴訟等の枠組みでの解決となります。
低分子医薬とそれ以外では対象として認められている類型の特許が異なるだけでなく、パテントリンケージ制度の下での取り扱いも異なるので(次号で説明)要注意です。
②ジェネリック申請の際の声明
特許で保護された新薬が承認され市場に出て時が経って、次はジェネリック薬の出番です。ジェネリック薬を販売するための承認を求めて薬事当局(NMPA)に申請(ジェネリック申請)する際、ジェネリック企業は当該ジェネリック薬に対応する特許情報プラットフォームに登録されている先発の新薬特許に関して声明を登録しなければなりません。それらの声明は、特許情報プラットフォームに登録されている新薬の特許が自己の申請するジェネリック薬とどのような関係にあるのかについての声明です。ジェネリック企業が提出することが求められている声明は下記の4種類です。(米国のPatent Linkage制度におけるParagraph IVの声明は、下記の声明IVに該当します。)
- 声明I:特許情報プラットフォームに関連する特許は存在しない。
- 声明II:特許情報プラットフォームに関連する特許は存在するが、すでに特許満了している、もしくは特許無効宣言されている。
- 声明III:特許情報プラットフォームに関連する特許は存在するが、ジェネリック企業はジェネリック薬が承認されたとしても当該特許が満了するまではジェネリック薬を上市しない。
- 声明IV:特許情報プラットフォームに関連する特許は存在するが、当該関連特許は特許無効が宣言されるべき、もしくはジェネリック薬は当該特許クレームの範囲には入らない。
ジェネリック企業が声明I~IIIを出した場合、申請に係るジェネリック薬に対応した新薬(先発品)をカバーする特許(新薬特許)が存在しないか、満了しているか、満了していなくても満了を待ってからジェネリック品を発売するということです。新薬の特許権者との間では特に特許の侵害問題は発生しません。
ところが、ジェネリック企業が声明IVを出した場合、ジェネリック薬をカバーする新薬特許が存在することは認めつつも新薬特許に挑戦(チャレンジ)するということです。これには新薬特許は無効だと主張する場合と、新薬特許がたとえ無効でなかったとしても(有効であったとしても)ジェネリック薬は新薬特許のクレームの範囲外にあると主張する場合が含まれます。もし声明IVが出されるような状況でNMPAがそのままジェネリック薬を承認して上市された場合には、上市後に特許侵害紛争が勃発するのは必至となります。かかる特許侵害紛争の勃発を事前に防ぐためにパテントリンケージ制度において、ジェネリック薬の申請段階で下記のメカニズムに従ってジェネリック薬の特許侵害問題の有無を判断し、もし発売したら特許侵害になるような場合には当該ジェネリック薬には上市の承認を付与しないというものです。
この声明はジェネリック申請が受理されてから10日以内に特許情報プラットフォームで公開されます(パテントリンケージシステムの実施弁法 §6)。
なお本制度の原案の段階では、ジェネリック企業が声明IVをNMPAに提出した際に対応する新薬承認取得者に対してこの声明IVの写しの送付を求めていませんでした。したがって新薬承認取得者はジェネリック申請を常にウォッチしておかなければならず、彼らの負担となることから問題視されていました。ところが今回の施行版では、ジェネリック企業は声明およびその根拠となる資料を新薬承認取得者に通知すること(その上で、新薬承認取得者が特許権者でない場合は新薬承認取得者は声明等を特許権者に通知すること)と規定されています。またジェネリック企業の声明が声明IVに該当する「ジェネリック薬は新薬特許クレームの範囲には入らない」である場合には、ジェネリック企業は声明の根拠となる資料としてジェネリック薬の技術と新薬特許のクレームの関係についての対比表、およびその関連資料の提出が必要となります(パテントリンケージシステムの実施弁法 §6)。
③裁判所・特許局への訴え
特許権者側によるジェネリック薬に対する45日以内の訴え提起
特許権者側はジェネリック企業が提出した声明IVの内容に異議がある場合、NMPAの審査部門(CDE)がジェネリック薬の申請がされたことを公示した日から45日以内に裁判所または特許庁に訴え出て、ジェネリック申請に含まれる技術内容(ジェネリック薬の物質、用途、製剤等の技術内容)が特許情報プラットフォームに掲載されている新薬特許クレームの範囲に入るか否かの判断を求めることができます(パテントリンケージシステムの実施弁法 §7)。
ここで訴え出ることができる特許権者側は特許権者に加えて利害関係人―当該特許権者から販売権等のライセンスを受けたライセンシーで、当該特許がカバーする新薬の上市の承認を取得している企業(新薬承認取得者) ― も同様に訴え出ることができます(最高裁規定 §2、特許庁弁法 §4(1))。
そして特許権者側は裁判所・特許庁が訴えを受理した日から15日以内にCDEとジェネリック薬の上市申請人に通知する必要があります(パテントリンケージシステムの実施弁法 §7)。
上記の期限内に裁判所・特許庁へ訴えが出されなかった場合CDEはジェネリック企業が提出した声明の内容に依拠して、ジェネリック薬の上市を承認するか否かを決定します(パテントリンケージシステムの実施弁法 §7)。
訴え出る先は裁判所か特許庁
訴え出る先が裁判所だけではなく、特許庁に対しても訴え出ることができることに違和感があるかもしれません。日本と違って中国の特許庁は特許侵害事件で侵害の行政裁定をする権限を有しています(特許法 §65、参照:特許行政 / 北京政府 VS 地方政府 知的財産権の侵害と行政救済)。一般の特許侵害訴訟で特許庁に訴え出る場合には当該特許局には地方政府の特許局も含まれますが、パテントリンケージ制度の下で訴え出ることができるのは国家知的財産局(北京の特許庁)です。特許庁は、担当部局として医薬品特許紛争早期解決システム行政裁決委員会を編成します(特許庁弁法 §2)。
裁判所に訴え出る場合は、北京の知的財産裁判所です(最高裁規定§1)。
裁判所への訴え
裁判所は特許権者側が先に特許庁に訴え出ていたとしても、並行して裁判所に訴え出ることを許容しています(最高裁規則 §5)。さらに裁判所は新薬特許の無効審判の請求が特許庁で受理されていることを理由に訴訟の中断の申し立てがなされたとしても、原則これを認めないとしています(最高裁規則 §6)。
また、裁判所において、被告のジェネリック企業が特許の無効を抗弁として主張はできませんが、被告が、①ジェネリック薬の技術は新薬特許の特許出願時に存在していた公知技術に含まれてる(公知技術の抗弁/特許法§67)又は、②ジネリック薬は新薬特許の出願時に既に事業化の準備等を進めていた(先使用権の抗弁/特許法§75②)との主張が認められ、裁判所は、かかる認定をした場合には、「ジェネリック薬は新薬特許のクレームに入らない」と判決を下すことが出来ます(最高裁規則 §7)。
特許権者等が保全の目的で、新薬特許の期間中、ジェネリック薬の製造・使用・販売・輸入の禁止を求めた場合、裁判所はかかる請求を審理の対象とするとしています。 然しながら、ジェネリック薬に関するNMPAへの申請行為・審査承認行為の禁止を求めたとしても、裁判所はこれを認めないとしています(最高裁規則 §10)。
尚、特許権者側が上記の45日以内に裁判所に訴え出なかった場合、ジェネリック申請者は、裁判所に対して「ジェネリック薬は、特許のクレームの範囲に入らない」ことの確認を求める訴訟を提起できます(最高裁規則§4)。
特許庁への訴え
特許庁に訴えることができるのは、特許権者、利害関係人(上記参照)、およびジェネリック薬の上市申請人です(特許庁弁法 §4(1))。
当事者がすでに裁判所に訴え出ている場合には特許庁への訴えは受理されません(特許庁弁法 §4(5),(6)、§10(9))。したがって裁判所と特許庁の両方に判断を仰ぐ場合には先に特許庁に裁定を求めていく必要があります。その後裁判所に訴え出ることは許容されています(裁判所規則 §5)。
また当事者が調停を望めば調停により解決を図ります。調停書が成立しない場合には特許庁は裁決に入ります(特許庁弁法 §15)。
裁決書にはジェネリック薬が新薬特許の保護範囲に入るか否かの認定、その理由・根拠が記載されます。そして裁決書はNMPAに回覧されると同時に公表されます(特許庁弁法 §18)。
当事者がかかる裁決に不服の場合には裁判所に訴えることが可能です(パテントリンケージシステムの実施弁法 §7、特許庁弁法 §19)。
その他
訴え出る際の必要な書類等についてですが、裁判所に訴える場合には最高裁規定§3に列挙、特許庁に訴える場合の書類は特許庁弁法§7,8に明示されています。もし書類不備等で不受理になった場合(特許庁弁法 §10)訴え出る45日の期限を過ぎてしまうと申し立ての機会を失うことになるので留意が必要です。特許庁で口頭審理がされる場合には5日前に場所・時間が通知されますが(特許庁弁法 §13)、短期間の事前通知なので対応の体制づくりが必要です。
特許権者側が特許庁と裁判所のどちらに訴え出るかは自由ですが、どちらが有利かそのプロス・コンスは別途に論じる予定です。
特許権者側が訴え出たとしてもCDAはジェネリック申請の審査を継続します。審査の結果技術的要件等を備えており承認できる状態になったとしても、訴え出た日からある一定期間ジェネリック承認が付与されません。この期間を待機期間と呼びます。次回はこの待機期間を中心に説明します。
④ 待機期間(化学医薬品)
⑤ 裁判所・特許庁の判断結果と薬事審査プロセスの関係(化学医薬品)
⑥ 声明IV以外のジェネリック申請(声明I, II、III)の取り扱い(化学医薬品)
⑦ チャレンジした最初のジェネリック薬⇒市場独占期間の付与(化学医薬品)
⑧ 化学医薬品以外(バイオ医薬、漢方)の取り扱い
⑨ ジェネリック薬の上市後の特許侵害訴訟
⑩ 法的責任:虚偽情報等の提出