以前紹介した「医薬品の特許紛争の早期解決システムの実施規則(药品专利纠纷早期解决机制实施办法)(試行)」の最終版が公表され、7月4日に施行されました。続いて、それに関連する裁判所、特許庁の細則も7月5日に公表されて施行され本格的に動き出すことになりました。
パテントリンケージ制度の全体像
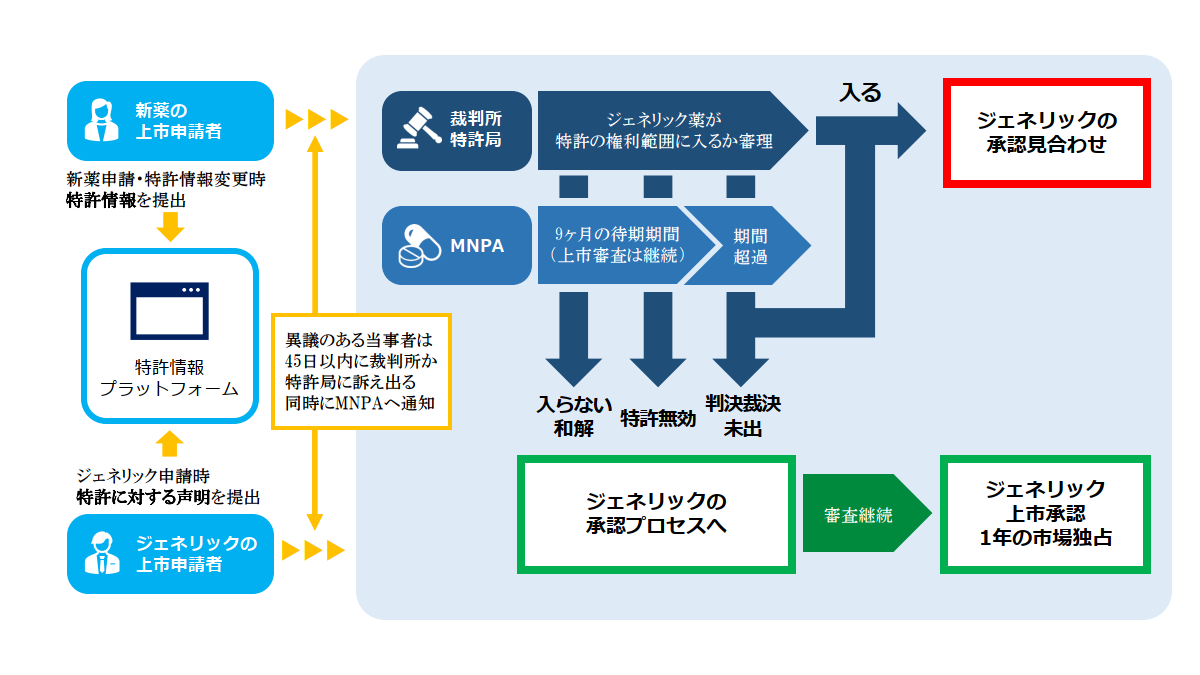
従来、ジェネリック企業があるジェネリック薬の上市の承認申請をした場合、中国の薬事当局はその審査段階においてジェネリック薬が医薬品として必要な基準を満たしているか否かの審査をするだけではなく、特許に関連する問題も判断していました。つまり、当該ジェネリック薬の製造・販売が対応の新薬(先発品)をカバーする新薬特許を侵害するか否かについてもある程度のレベルで判断がされていて、問題がないと判断されてはじめてそのジェネリック薬に対して上市の承認が付与されていました。そこには外部の様々な利害関係者からの圧力がかかっていたであろうことは想像に難くありません。そして今回のパテントリンケージ制度では、特許問題の判断にあたっては薬事当局がジェネリック薬の承認審査段階で単独で行うのでなく、裁判所、特許局とも緊密な連携を取って早期に解決しようというものです。
したがって特許法76条に定められているパテントリンケージ制度については、上記の3つの機関がそれぞれ下記の通り細則・弁法等を公表した上で施行されています。
- 薬事当局・特許庁の連名:<医薬品の特許紛争の早期解決システムの実施弁法(药品专利纠纷早期解决机制实施办法(试行))>(以下、パテントリンケージシステムの実施弁法)
- 最高裁:<ジェネリック薬の承認申請の審査中に発生した特許民事事件の法律適用についての若干の問題に関する規定(关于审理申请注册的药品相关的专利权纠纷民事案件适用法律若干问题的规定)>(以下、最高裁規定)
- 特許庁:<医薬品特許紛争の早期解決システムにおける行政裁決に関する弁法(药品专利纠纷早期解决机制行政裁决办法)>(以下、特許庁弁法)
これらの弁法・規定を引用しながら、下記の全体像に従って説明を進めて行きます。
特許情報プラットフォームの開設
中国の薬事当局である医薬品監督管理局(NMPA)の審査センター(CDE)がパテントリンケージ制度の肝となる、新薬とそれにリンクした特許を収録した特許情報プラットフォーム(https://zldj.cde.org.cn/home)を開設しその管理責任を負います(パテントリンケージシステムの実施弁法 §2,3)。特許情報プラットフォームはアメリカのオレンジブックの中国版と言えます。
パテントリンケージ制度でのプロセスは以下の通りです。
①新薬の特許情報の提供
②ジェネリック申請の際の声明
③裁判所・特許局への訴え
④ 待機期間(化学医薬品)
パテントリンケージシステムの実施弁法§8
1)待機期間の内容
特許権者・利害関係人は化学医薬品ジェネリック申請の際に提出された声明IVに異議がある場合に、裁判所または特許庁に訴え出ることができます(前述③を参照)。特許権者等が45日以内に訴え出た場合、NMPA(医薬品監督管理局)はジェネリック申請に対して9ヶ月の待機期間を設けて、その間はジェネリック申請に対して承認を付与しないことになります。 特許権者等が45日以内に訴え出なかった場合、NMPAは通常の審査プロセスを経て薬事要件を満たしていれば、ジェネリック申請に対して上市承認を付与します。 なお待機期間の起算日は裁判所又は特許庁が特許権者等から訴えを受理した日です。また待機期間は一回のみ設定されます。
この待機期間の性格ですが、この間であってもCDE(NMPAの審査部門)はジェネリック申請に対して審査は継続されますが、上記の通りたとえジェネリック申請が薬事要件を満たしていたとしてもその間は上市の承認は付与されないことになります。
2)米国との比較
米国のMarket ExclusivityおよびPatent Linkage制度の下では、ジェネリック企業は先発の新薬が承認されてから4年経過後にジェネリック申請(ANDA)をする事ができ、その際Paragraph IV(中国の声明IVに該当)の声明を提出している場合に特許権者等は訴訟の提起が可能となり、そのときの待機期間は2年半となっています。したがって先発の新薬は、訴訟の提起を前提として少なくとも4+2年半=6年半は独占権を保証されており、この間にジェネリック薬が上市されることはありません。これに対して、中国はMarket Exclusivityに該当するデータ保護制度の導入は検討されている段階です。したがって現段階では先発の新薬が承認されれば、ルール上いつでもジェネリック申請が可能であって、声明IVが付されていた場合に特許権者が訴え出たとしても9ヵ月間しかexclusivityが保証されていないということになります。いずれにしてもデータ保護制度の導入を待っている段階です。
3)待機期間=化学医薬品に対してのみ設定
この待機期間は、化学医薬品についてのみ設定されています。したがってバイオ医薬品、漢方薬については待機期間はなく、ジェネリックが申請された場合には通常の審査プロセスを経て承認されます。ジェネリック薬の上市後に通常の侵害訴訟によって特許問題を解決して行くことになります。
⑤ 裁判所・特許庁の判断結果と薬事審査プロセスの関係(化学医薬品)
パテントリンケージシステムの実施弁法§9
化学医薬品のジェネリック申請で声明IVが提出され、これに対して特許権者等が異議ありとして裁判所・特許庁に訴え出た場合、上記の通り9カ月の待機期間が設定されます。この待機期間中に裁判所・特許庁から判断(裁判所=判決、特許庁=裁定)が下りた場合、特許権者等は当該判断(判決・裁定)を10日以内にCDEに通知する必要があります。CDEはジェネリック申請に対して下記の通り審査・処理します。
1)判決・裁定が下記①、⑤の内容の場合⇒新薬特許の満了を待って、CDEはジェネリック申請の承認手続きに入る。
① ジェネリック薬は新薬特許の範囲内に入る。
⑤ 待機期間を経過後であっても、ジェネリック申請の上市承認の付与前に、ジェネリック薬は新薬特許の範囲内に入るとの判決・裁定が下りた場合。
ただし上記の場合であったとしても、その後下記の事象が発生した場合、ジェネリック申請の企業はNMPAに対してジェネリック薬の上市承認を付与するよう請求することができる。そのような場合には、NMPAは承認を付与するか否かを決定することができる。
① 判決・裁定が次の裁判手続きで覆った場合
② ジェネリック企業と特許権者等の間で和解が成立した場合
③ 新薬特許が無効との判断が下りた場合、または、
④ 特許権者等が訴訟・裁定請求を取り下げた場合
2)判決・判定が下記②~④の場合⇒新薬特許の満了を待たずにCDEはジェネリック申請の承認手続きに入る。
② ジェネリック薬は新薬特許の範囲に入らない。
③ 新薬特許は無効
④ 9ヶ月の待機期間内に、判決・裁定が下りない。
⑥ 声明IV以外のジェネリック申請(声明I, II、III)の取り扱い(化学医薬品)
パテントリンケージシステムの実施弁法§10
「②ジェネリック申請の際の声明」で列挙した声明I(特許情報プラットフォームに関連する特許は存在しない)または声明II(特許情報プラットフォームに関連する特許は存在するが、すでに特許満了している、もしくは特許無効宣言されている)が提出されている場合、NMPAは当該ジェネリック申請に対して技術的な審査の結果を踏まえて上市の承認をするか否かの判断を下します。
また声明III(特許情報プラットフォームに関連する特許は存在するが、ジェネリック企業はジェネリック薬が承認されたとしても当該特許が満了するまではジェネリック薬を上市しない)が提出されている場合、NMPAは、同様な方式で判断を下します。但し承認が付与されたとしても、当該ジェネリック薬は、特許期間の満了を待って、上市が可能となります。当該ジェネリック申請より前に申請していたジェネリック薬が声明IVを提出し、それが認められて、後記の通り市場独占期間が付与されている場合には、当該独占期間の満了後に、当該ジェネリック申請に上市承認が付与されます。
⑦ チャレンジした最初のジェネリック薬⇒市場独占期間の付与(化学医薬品)
パテントリンケージシステムの実施弁法§11
ジェネリック企業が声明IVを出してジェネリック薬をカバーする新薬特許が存在することは認めつつも新薬特許に挑戦(チャレンジ)してその主張が認められ、さらにNMPAがジェネリック申請に対して上市承認を付与した場合には、最初の承認対象となったジェネリック薬に対して1年間の市場独占期間が与えられます。
つまり新薬特許にチャレンジしたジェネリック企業に褒美を挙げるという趣旨で、このあと1年間は2番目以降のジェネリック薬の上市承認を与えませんので、最初の1番目のジェネリック薬にはジェネリック薬市場での1年間の独占期間が与えられることになります。ただしこの1年間の市場独占期間は新薬特許の有効期間を越えては付与されません。
ここで新薬特許にチャンレンジが認められるとは、ジェネリック申請が声明IVを提出しており、かつジェネリック申請者が特許庁に対して新薬特許の無効審判を請求し、審査の結果無効の判断がされ、最終的にNMPAによってジェネリック申請が上市承認されることを言います。
前述の待機期間と同様、最初のジェネリック薬に付与される1年間の市場独占期間中であっても2番目以降の他のジェネリック申請に対してはCDEは技術審査を継続することができます。したがって他のジェネリック申請は市場独占期間が満了すれば承認付与のプロセスに入ります。
米国の制度の下では最初のジェネリック薬に180日間の独占期間が付与されるのに対して、中国では1年間です。中国の場合ジェネリック薬が承認されたとしても市場に浸透するまでに時間がかかることが一つの理由として挙げられています。
⑧ 化学医薬品以外(バイオ医薬、漢方)の取り扱い
パテントリンケージシステムの実施弁法§12、13
バイオ医薬と漢方については新薬承認取得者による新薬特許情報プラットフォームへの登録(パテントリンケージシステムの実施弁法§2, 3, 4)および新薬特許権者側による裁判所・特許庁への訴え(パテントリンケージシステムの実施弁法§7)が適用されます。 しかしながら同実施弁法§8の適用がないことから、裁判所・特許庁に訴え出ても待機期間は設定されない事になります。バイオ医薬と漢方について新薬特許情報プラットフォームに登録できる特許の類型は化学医薬品と異なっています(前述①参照)。
バイオ医薬と漢方薬のジェネリック薬(バイオシミラー等)の上市申請が出された場合、NMPAは技術審査を実施後に要件を満たしていれば承認の決定を下します。ただし裁判所・特許庁が当該ジェネリック薬が新薬特許の範囲内に入るとの決定を下した場合には当該ジェネリック薬は新薬特許の有効期間の満了後に上市が可能となります。
⑨ ジェネリック薬の上市後の特許侵害訴訟
パテントリンケージシステムの実施弁法§14
ジェネリック薬(化学医薬品、バイオ医薬、漢方)が上市承認された後に特許権者等が当該ジェネリック薬は新薬特許を侵害していると判断する場合には、特許法等の下で特許侵害訴訟の提起等によって紛争を解決することが可能です。ただしその場合であってもジェネリック薬に付与された上市承認の取消等の効果は発生しません。
⑩ 法的責任:虚偽情報等の提出
パテントリンケージシステムの実施弁法§15、最高裁規定§8、9、特許庁弁法§20、21
ジェネリック企業がジェネリック申請の際に提出する声明の内容に虚偽がある場合、もしくは特許権者が故意に新薬特許情報プラットフォームに無関係の特許等を登録する等の行為によって相手方当事者に損害が発生した場合、さらには裁判所・特許庁に提出した証拠等に虚偽がある場合、または相手方の秘密情報を漏洩した場合等、法的な責任を負うことになります。
以上、中国のパテントリンケージ制度の概観です。今後の実務等を通じで不明点が解消されて行くことになると思います。一定期間を置いて更に解説をする予定です。