中国薬事制度改革(第1回)― 激動の 2015 年
1.始めに
CFDA(中国の国家食品薬品監督管理総局)は、2015 年の年頭に、今年を医薬品審査承認制度の改革元年にするとして、業界内に風雲急を告げる年が始まりました。その後、ご承知の通り、次々と関連する文書・通知が発せられましたが、その重要なものは下記の通りです。
| 公布日 組織名 |
文署名 | 主要な内容 |
| 2015.08.18 国務院 |
医薬品・医療機器の審査承認制度の改革方針 (国発国发〔2015〕44 号) |
・新薬の定義の変更 ・NCE 新薬の審査承認のスピード化 ・販売承認制度への移行 ・海外未承認薬について、中国国内での臨床試験の実施に関する承認システム |
| 2015.07.31 CFDA |
承認申請の滞貨問題の解決の為の政策意見 (140 号通知) |
・ジェネリック薬の水準向上 ・IND(臨床試験申請)の審査承認の最適化(届出制) ・臨床上、必要性が高い医薬品の承認 |
| 2015.01.30 CFDA |
国際共同治験ガイドライン(試行)の通知 (2015 年第 2 号) |
・国際共同治験について、総合的な実施要件 ・規範性・科学性面での要件、登録申請の要件、プロトコール等の変更等に関する手続・管理面での要件 |
本論文では、2015 年に国務院及び CFDA が公布した重要な通達等の全体を俯瞰して、下記のようなテーマにて、将来の新しい審査承認制度の方向性を分析し、我々業界人のビジネスにどのような影響を与えることになるのかについて検討していきたいと思います。
<第一回>
1.始めに
2.「医薬品・医療機器の審査承認制度の改革方針」(44 号-改革方針)
(1) 新薬の定義の変更
(2) 新薬の研究開発の強力な推進
(3) 国際共同治験のデータの受け入れ
<第二回>
(4)医薬品の販売承認制度の試行
(5)グローバル臨床開発とのシンクロナイズ
3.「承認申請の滞貨問題の解決の為の政策意見」
4.国際共同治験ガイドライン(試行)の通知
5.その他の重大政策
<第三回以降>
その時点での立法動向を踏まえて、テーマを設定予定。
本レポートにより、新薬(NCE)に関する規制動向を中心に、日本の医薬品業界の方々が中国の薬事法
規の理解を深められることに資すれば幸いです。
2.医薬品・医療機器の審査承認制度の改革方針
中国国務院は、2015 年 8 月 18 日に「医薬品・医療機器の審査承認制度の改革方針」を発表し、医薬品の審査・承認の改革に関し、5 項目からなる目標を設定し、12 項目の主要任務及び 4 項目の保障措置からなる改革方針を打ち出しました(国発(2015)44 号 / 以下「44 号-改革方針」)。新薬(NCE)に関する重要なポイントは、下記に示す通りです。
(1)新薬の定義の変更
1)定義変更の具体的な内容
現行の「医薬品登録管理弁法」によれば、中国国内で未だ発売・販売されていない医薬品の申請を、「新薬」申請と呼ぶと定義されています。改正後は、「新薬」は、中国の国内外で未だ発売・販売されていない医薬品であること、そして、新薬のオリジナリティ・新規性に基づいて、「創新薬(イノベーション薬)」と「改良型新薬」の二つに分類されるとしています。(末尾の分類表 1 及び 2)尚、ジェネリック薬については、現行の「既に国家標準薬とされている薬剤を模した医薬品」から改正後は「先発品(研究開発特許品)を模し、先発品と品質・治療効果が一致する医薬品」とされます。(末尾の分類表 3〜5)
2)予想される影響
- 現行制度の下での趨勢
現行法の下では、海外で発売されていたとしても、中国国内で未だ発売されていない薬剤は、3.1類の「新薬」と呼ばれています。これは、例えば、米国でオリジナル企業が各種試験を実施して新薬の安全性・有効性等のデータを取得の上、FDA に申請後、承認が下りた段階で、情報公開法の下で、FDA に申請された主要データが公開されます。この新薬について、中国で特許保護がされていない等の背景がある場合には、中国企業が公開情報に基づいて、当該医薬品を新薬として開発し、申請・承認取得の上、販売していました。このような 3.1 類の新薬に対しても、3 年間の新薬監測期間(日本の再審査期間に該当)が設定され、この 3 年間の間、当局は、第三者からの医薬品の申請(輸入申請、中国国内申請)を認めません。この 3 類の新薬は、海外の新薬について中国で最初に模して開発したところに意味がありますが、非常に沢山の申請がされており、ジェネリック薬(6 類に分類される)に比べても、下図の比較表から見て明らかなとおり、申請数が多いとされています。(2015 年 8 月 1 日の中国医薬品審査センターの公表数字による)
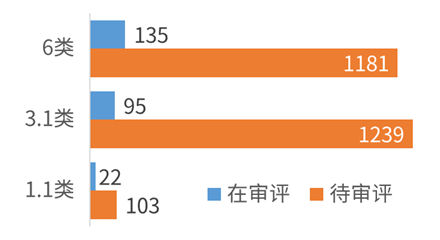
<注>
6 類:ジェネリック薬、
3.1 類:海外で発売済、中国国内で未発売の「新薬」
1.1 類:中国の国内外で未発売の「新薬」(NCE 及びその製剤品) - 改正後の姿
新薬と呼ばれる範囲を大幅に減縮することに繋がる今回の新薬の定義の改正に伴い、従来からの医薬品の登録分類(1類〜6 類からなる分類)も見直しがされることになっています。医薬品は、「創新薬(イノベーション薬)」、「改良型新薬」及び「ジェネリック薬」の三つの概念に分かれることになります。現在、「医薬品登録管理弁法」の改正作業が進んでいますが、その中で、分類の新しい枠組みが示されることになり、改正動向を注目していく必要があります。この様な政策面での転換期を迎え、従来の 3.1 類新薬の申請に我先に走るといった傾向が密かに進行しています。 改正により従来の 3 類の新薬としての位置づけがなくなった場合には、これまで、ある種の独占保護の期間として与えられていた「監測期間」が自ずとなくなることを意味します。
従来の 3 類が今後はジェネリック薬のカテゴリーに入ることになり新薬としてのビジネスチャンスもなくなることになります。従来の 3 類のような新薬に与えられていた政策的な優遇措置の適用も受けられなくなり、今後、中国では所謂「新薬」に分類される薬剤の数が急速に減っていくことになると思われます。尚、2015 年 11 月 6 日、CFDA は、「化学医薬品の登録分類に関する改革プラン」を公表し、パブリックコメントを求めております。そのプランによりますと、下記のような分類が現在、検討の俎上に上っています(従来とは分類番号が変わっているので要注意)。新登録分類
(検討段階)分類の基準 現行の旧分類 1 中国の国内外で未だ発売されていない創新薬(イノベーション新薬) 1 2 中国の国内外で未だ発売されていない改良型新薬 1, 2, 4, 5 3 海外で発売済、中国で未発売の薬剤についてのジェネリック薬 3 4 中国で発売済の薬剤についてのジェネリック薬 6 5 海外で発売済の薬剤について、中国で販売の為の申請をする薬剤
(オリジネータ―又はライセンシーによる申請)輸入医薬品 - 分析
武田薬品の新薬である 2 型糖尿病治療剤(Trelagliptin succinate / DDP-4 阻害剤)を例にとって考えたいと思います。本剤は、2015 年 3 月に日本で承認されております。中国では、物質特許が存在しませんが、適応症に関する用途特許(WO2007033350、CN101374523B / Dipeptidyl peptidase inhibitors for treating diabetes)があり、その特許期間は、2026 年 9 月 13 日まであるとされています。10 数年の用途特許の残存期間があるにもかかわらず、日本で発売後、半年を経たずに、中国では 12 社の中国の医薬品企業が 3.1 類の新薬の申請をしています。
従来、中国の医薬品企業が新薬の申請をした際、CFDA は通常、当該新薬に関わる申請者(上記の例で言えば、武田薬品)の物質特許の存在のみを重要視しており、適応症に関する用途特許については考慮に入れてなかったとされます。このように中国の企業が申請した従来の 3.1 類の新薬は、将来どのように処理されるのでしょうか? 法改正が行われる前に既に申請済みの 3.1 類の新薬は、現行の従来規則に従って、審査の上、承認が与えられるとされています。然しながら、具体的にどのような取り扱いがされるのか、今後、正式に公布される実施規則等の制定を待つ必要があります。
(2)新薬の研究開発の強力な推進
1)新薬の創出に向けて、「44 号-改革方針」では、新薬の審査・承認の手続きの簡素化を打ち上げており、新薬の上市をより早める方向性が明確に示されています。これに対して、ジェネリックは、業界で大地震の発生が予見される様相を呈しています。中国の医薬品企業は、ジェネリックを事業の柱としているわけですが、それらの多くは淘汰される局面に立たざるを得ない状況です。
2)予想される影響
- 現行制度の下での趨勢
中国は、正に、ジェネリック薬の大国、それは、医薬品の総使用量の 80%をジェネリックが占めている事実に現れています。これまで中国で生まれた新薬は日米欧に比較して非常に少ないわけですが、今年のノーベル生理学・医学賞の対象となった抗マラリヤ薬、アーテミシニンは、1970 年代の発明で、中国では 1980 年代から使用され始めていました。中国発の新薬が 30 数年を経てやっと世界に認知されたといった状況です。中国発の新薬自体が少ないこともありますが、行政面では、審査の遅延問題が発生しています。1.1 類の NCE の場合、IND 申請から製造承認までの平均期間が 7.5 年とされていますが、その期間中の少なからざる時間が、当局への申請後の審査待ちの時間とされています。 - 改正後の姿
近年、中国は正に新薬の創生に向けて様々な施策を打ってきています。「44 号改革方針」では、新薬の審査に当たって、特殊審査制度を充実させるとしており、審査・承認の期間が短縮化される対象の薬剤として、エイズ・癌・重大な感染症・希少病等の疾病の新薬、国の重大科学技術プロジェクト・国家重点 R&D 計画の対象となっている医薬品、中国での国内生産に切り替えられた新薬、小児用薬、並びに先進的な製剤技術・画期的な治療手段を用い、又は治療効果が顕著に表れている新薬が挙げられています。 - 分析
「44 号改革方針」は、CFDA(中国の国家食品薬品監督管理総局)ではなく、国務院からの発表となっていることからも、中国に於ける新薬 R&D に対する重点化は、既に、国家戦略の段階に達しており、中国での新薬の創出は、新しい発展段階を迎えていると言えます。中国での新薬 R&D の環境を整備することにより、イノベーションを更に促進し、中国の製薬企業の R&D の積極性を引き出すことに資しています。中国国内での新薬の水準を引き上げ、更には、早期に新薬の発売に持って行くことが狙いにあります。
(3)国際共同治験のデータの受け入れ
1)これまで、中国での国際共同治験の実施について審査・承認上の問題が発生したことにより、多くの多国籍企業は、中国での国際共同治験の実施に興味を失っていく趨勢にありました。ところが、今回の「44 号改革方針」では、制度設計上の意味からも、海外との同時開発を実現する為に国際共同治験を奨励する立場を取っており、中国の国内外の研究開発企業に対し、国際共同治験の推進を促し、新薬の上市の短縮化に繋げたいとしています。また、「44 号改革方針」では、ある一定の要件を満たしていることを条件に、国際共同治験に基づく臨床試験データを中国での承認申請に使用できると明確に述べています。海外の R&D 企業が海外で生産した新薬に関する輸入医薬品についても、開発段階で中国を組み込んだ形で国際共同治験を実施することにより、中国での上市までの時間が短縮化される政策的な根拠となるので、輸入医薬品の上市のスピードアップ化が確実に期待される状況となりました。
2)予想される影響
- 現行制度の下での趨勢
これまでは、中国では、中国内資企業を保護するとの考え方に基づき、所謂、三申三承(IND とNDA の二度の申請と二度の承認でなく、3 度の申請と 3 度の承認)等の政策手段をとることによって、海外企業が新薬の輸入承認を取得するまでの時間の引き延ばしを図ってきたとも言えます。中国では新薬の承認が海外に比べ、3〜5 年の遅れをきたし、甚だしい場合には、10 年後に中国で臨床での使用が開始されるようなケースもありました。 - 改正後の姿
「44 号改革方針」で打ち出されている新政策では、審査承認の大幅な短縮化という意味でも、大鉈が振るわれ、特に、海外企業の開発にかかわる輸入医薬品については、承認までの時間が短縮されることが期待されています。ここで、見過ごしてはならないことは、中国が国際共同治験を推進ということは、中国の内資であって新薬の R&D を行う企業にとっても強く有利に働くということです。即ち、中国の新薬 R&D 企業が、例えば、国際展開のすすんでいる中国系の CRO(臨床試験の受託機関)を巻き込んだ形での連携によって、国際共同治験を実施し、中国発の新薬の国内外での上市を早めるという方向に展開するということです。 - 分析:「アジア太平洋地域で先行させる複線モデル」
中国の新薬 R&D 企業の和記黄埔医薬(Hutchison 社)が、自社の R&D から生まれた抗癌剤(Volitinib)について、アストラゼネカ社との開発の提携を公表したのは、4 年前の 2011 年 12 月のことでした。これは、中国の企業が臨床段階に入った低分子化合物の医薬品を多国籍企業にライセンスし、提携関係にはいった本格的な第一号でした。Hutchison 社は、2012 年 2 月に豪州で Phase I の臨床試験を開始すると同時に、中国では、CFDA に IND を申請しました。そして、翌年 6 月に中国で Phase I を開始。中国の臨床試験の計画・実施を豪州の Phase I と関連付けたことから、中国での臨床開発を大幅に短縮させることが出来ました。このように海外の臨床試験データを利用し、また、開発時間を短縮することにより、中国での上市の早期化に繋げることが可能となります。豪州以外にも、韓国、台湾等を含めた「アジア太平洋で先行させる臨床開発の複線モデル」によって、中国での臨床開発の展開の可能性を広げることが出来るようになってきています。新薬の臨床開発の初期段階で中国での開発とアジア太平洋地域の開発を連携させることにより、中国での臨床試験の立ち上げの早期化を図り、その後、国際共同治験の実施に繋げていくことが、周辺地域を含めた中国での上市の早期実現を図る道と考えられます。
以上
投稿者プロフィール
最新の投稿
 政策行政2016年9月1日中国 / 医薬品の承認審査に関する法規制の現状
政策行政2016年9月1日中国 / 医薬品の承認審査に関する法規制の現状- 医薬市場2015年12月27日中国薬事制度改革(第2回)― 激動の 2015 年
- 医薬市場2015年11月23日中国薬事制度改革(第1回)― 激動の 2015 年
トラックバック & ピングバック
[…] 中国の2015年は、「医薬品審査承認制度」の「改革元年」として位置づけられると思います。今年、下記のリストに示された重要な政府通知が発せられました。今回の中国医薬品ビジネス・レポートNo.2では、前回に引き続いて、「改革の方向性」、「承認申請の滞貨問題」及び「国際共同治験のガイドライン」に関するポイントを採り上げます。 […]
返信を残す
Want to join the discussion?Feel free to contribute!